
Gilbert syndrome (OMIM 143500) is a hereditary hyperbilirubinemia in which the concentration of total and unconjugated bilirubin in the blood is increased.
Gilbert syndrome usually occurs in 3.0-13.0% of the population.
orderSchmid (1995) pointed out that Gilbert syndrome is completely benign and clinically insignificant, requiring neither treatment nor long-term medical monitoring.
Because the diagnosis is largely a diagnosis of exclusion, clinicians sometimes find it difficult to allay lingering concerns about serious liver disease, causing patients unwarranted anxiety.
Normally, indirect bilirubin (a toxic breakdown product of hemoglobin) should be converted into non-toxic direct bilirubin and excreted with bile.
BUT with a mutation, there is a suppression of an enzyme that converts indirect bilirubin to direct bilirubin, and bilirubin accumulates in the body.
This leads to an increase in bilirubin in the blood (usually not more than 80-100 µmol/l, with a significant predominance of indirect bilirubin) and to the occasional occurrence of moderate jaundice (staining of the skin, mucous membranes, whites of the eyes yellow). Other biochemical blood parameters and liver tests remain within normal limits.
Gilbert syndrome is more common in men (2-3 times more common than in women) and usually first appears at the age of 3-13 years. It is inherited by autosomal recessive type.
THE CAUSE OF GILBERT SYNDROME
UGT1A1 GENE
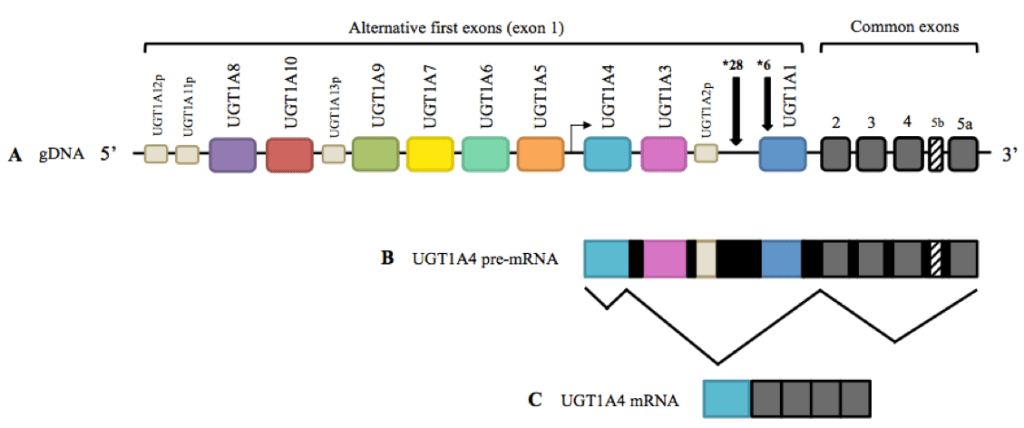
Changes in the nucleotide sequence of the UGT1A1 gene are the cause of changes in the UDF-glucuronosyltransferase enzyme activity in SJ, SCNI, and SCNII.
The UGT1A1 (UDP glucuronosyltransferase family 1 member A1, 2q37.1) gene encodes uridine diphosphate-glucuronosyltransferase 1A1, which belongs to the conjugation enzyme superfamily
2022. A. Ivanova, V. N. Maksimov. Molecular genetic aspects of Gilbert's syndrome, Crigler- Najjar syndromes types I and II Experimental & clinical gastroenterology, No 204 (8) 2022.
rs3064744
The most common molecular genetic diagnosis in CS is the determination of the number of TA repeats of the UGT1A1 gene promoter (rs3064744, c.-54_-53insAT).
The most frequent variant in the gene promoter in the population is A(TA)6TAA (UGT1A1*1).
An increase in the number of TA repeats, most commonly up to 7 - A(TA)7TAA (UGT1A1*28) was found in CJ. A (TA)5TAA (UGT1A1*36) and A(TA)8TAA (UGT1A1*37) variants are also found in the population.
The frequency of promoter carriage with an increased number of repeats in the Caucasoid population is about 30-40%.
About 16% of the Caucasian population are homozygous carriers of the A(TA)7TAA variant, and only half of them are diagnosed with clinical symptoms of CSF, which is associated with low penetrance and expression of the variant.
A large Russian study has shown that an increase in the number of TA repeats of the gene is associated with increased levels of total and unconjugated bilirubin, including carriage of rare allelic variants (TA≤5, TA≥8).
A(TA)5TAA, A(TA)7TAA, and A(TA)8TAA variants are associated with 130, 65, and 50% enzyme activity UDF-glucuronosyltransferase 1A1, respectively, compared with the A(TA)6TAA variant.
A decrease in expression of the UGT1A1 gene due to an abnormality in the promoter region is thought to be a necessary but not sufficient change for the development of clinical manifestations of CS.
To date, CSF is one of the monogenic diseases whose development is caused by changes in the nucleotide sequence of a single gene.At the same time, incomplete penetrance and variable expression of clinical manifestations of the rs3064744 variant of the UGT1A1 gene are observed, which may be associated with other molecular genetic markers that may contribute to the development of the syndrome.
Thus, it is likely that SJ is not monogenic but oligogenic syndrome!
2022.A. Ivanova, V. N. Maksimov. Molecular genetic aspects of Gilbert's syndrome, Crigler- Najjar syndromes types I and II // Experimental & clinical gastroenterology, No 204 (8) 2022.
Variants in the UGT1A1 gene are associated with the development of familial transient hyperbilirubinemia in newborns and metabolism of several drugs.
Thus, in the case of UGT1A1*6, rs3064744 gene variants, administration ofindinavir, irinotecan, atazanavir, sorafenib, tocilizumab, belinostat and paracetamol (acetaminophen) is associated with increased toxicity.
rs4148323 (UGT1A1*6)
The rs4148323 (UGT1A1*6) variant is associated with an increased risk of neonatal hyperbilirubinemia; in the homozygous form, the variant is associated with the development of SJ.
According to gnomAD variant is more common in East Asia (the frequency of the rare allele in the Asian population is about 0.15, in the European population - about 0.05).
According to RUSeq the variant frequency in the Russian population is about 0.014.
The UGT1A1*6 variant is associated with a 70% reduction in UDF-glucuronosyltransferase 1A1 enzyme activity compared with the wild type.
In a large population study in China,the A(TA)7TAApromoter variant and the A allele of the UGT1A1*6 variant of the UGT1A1 gene were shown to be associated with elevated total bilirubin levels .
In a recent study in China, molecular genetic analysis of the promoter and exons of the UGT1A1 gene was performed in 120 individuals with CSF and 120 healthy control group participants.
According to the results of the study, there were more UGT1A1*6 and UGT1A1*28 heterozygotes than in the study in India - 20.83%.
The homozygous carriers of the UGT1A1*28 variant in the group with CS were 20% and 15% of the UGT1A1*6 variant.
The level of total bilirubin was higher in homozygous carriers of the UGT1A1*28 variant than in carriers of other variants of the UGT1A1 gene nucleotide sequence in the group with CSF.
2022.A. Ivanova, V. N. Maksimov. Molecular genetic aspects of Gilbert's syndrome, Crigler- Najjar syndromes types I and II // Experimental & clinical gastroenterology, No 204 (8) 2022.

SYMPTOMS
- Complaints of heaviness in the right subcostal area
- fatigue
- a bitter taste in the mouth
- nausea
- vertigo
- Jaundiced sclerae and skin. (The skin and sclerae turn yellow when blood bilirubin levels rise. This is the most alarming symptom).
⠀
ASIAN MUTATION

Asian mutation - G71R replacement of glycine with arginine (Gly71Arg).
Since 2011, more than 4,500 people with suspected Gilbert Syndrome have come to us. After analyzing statistics and feedback from physicians, our team began to study the Asian Mutation, as Asians were twice as rarely found with the corresponding clinical picture.
Method validation and testing of several patients with obvious symptoms of SJ, in whom the classical TA7 mutation was not found, showed the presence of Asian G71R.
So we added the Asian mutation to our genetic analysis a Gilbert Syndrome.
DESCRIPTION
- Suspicion of Gilbert syndrome;
- Differential diagnosis of Gilbert syndrome, and other diseases manifested by hyperbilirubinemia;
- Before treatment, with the use of drugs with hepatotoxic effects;
- Mild infectious jaundice;
- Chronic jaundice;
- Increased bilirubin concentration with other normal biochemical blood parameters;
- A history of a poor family history (non-infectious jaundice, hyperbilirubinemia).
⠀
TREEGENE LAB STATISTICS