
Синдром Жильбера (OMIM 143500) — наследственная гипербилирубинемия, при которой происходит увеличение концентрации общего и неконьюгированного билирубина в крови.
Синдром Жильбера обычно встречается у 3,0–13,0% населения.
заказатьSchmid (1995) указал, что синдром Жильбера является полностью доброкачественным и клинически незначимым явлением, не требующим ни лечения, ни длительного медицинского наблюдения.
Поскольку диагноз в значительной степени является диагнозом исключения, клиницистам иногда бывает трудно развеять затянувшиеся опасения по поводу серьезного заболевания печени, что вызывает у пациентов необоснованное беспокойство.
В норме непрямой билирубин (токсичный продукт распада гемоглобина) должен превращаться в нетоксичный прямой и выводиться с желчью.
НО при мутации, идет подавление фермента, который и переводит непрямой билирубин в прямой, и билирубин накапливается в организме.
Это приводит к повышению билирубина в крови (как правило, не более 80-100 мкмоль/л, со значительным преобладанием непрямого билирубина) и к периодическому возникновению умеренной желтухи (окрашивания кожи, слизистых оболочек, белков глаз в желтый цвет). Остальные биохимические показатели крови и печеночные пробы остаются в пределах нормы.⠀
Синдром Жильбера чаще встречается у мужчин (в 2-3 раза чаще, чем у женщин) и обычно впервые проявляется в 3-13 лет. Наследуется по аутосомно-рецессивному типу.
ПРИЧИНА СИНДРОМА ЖИЛЬБЕРА
ГЕН UGT1A1
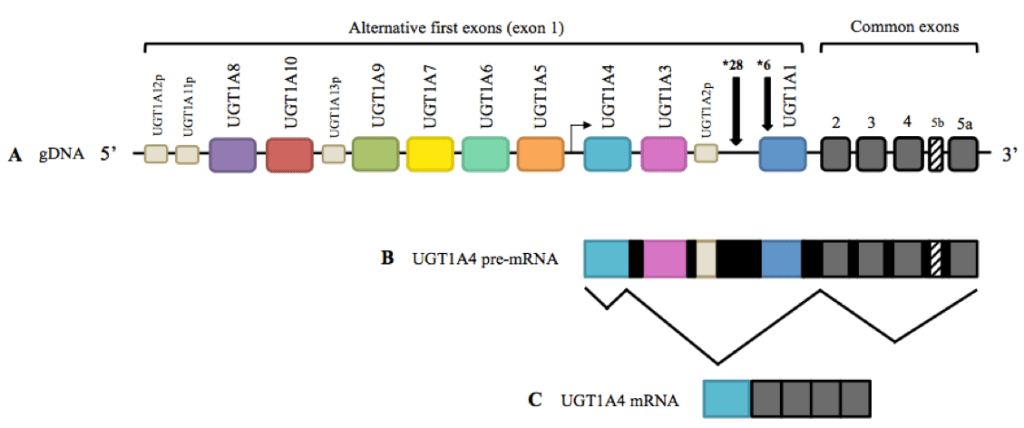
Причиной изменения активности фермента УДФ-глюкуронозилтрансферазы при СЖ, СКНI, СКНII являются изменения нуклеотидной последовательности гена UGT1A1.
Ген UGT1A1 (UDP glucuronosyltransferase family 1 member A1, 2q37.1) кодирует уридиндифосфат-глюкуронозилтрансферазу 1A1, относящуюся к надсемейству ферментов конъюгации
2022. A. Ivanova, V. N. Maksimov. Molecular genetic aspects of Gilbert's syndrome, Crigler- Najjar syndromes types I and II Experimental & clinical gastroenterology, № 204 (8) 2022.
rs3064744
Наиболее распространенной молекулярно-генетической диагностикой при СЖ является определение количества ТА повторов промотора гена UGT1A1 (rs3064744, c.-54_-53insAT).
Наиболее частый вариант в промоторе гена в популяции – A(TA)6TAA (UGT1A1*1).
При СЖ было найдено увеличение количества ТА повторов, чаще всего до 7 – A(TA)7TAA (UGT1A1*28). Также в популяции встречаются варианты A(TA)5TAA (UGT1A1*36) и A(TA)8TAA (UGT1A1*37).
Частота носительства промотора с увеличенным количеством повторов в европеоидной популяции около 30–40%.
Около 16% европеоидной популяции являются гомозиготными носителями варианта A(TA)7TAA, и только у половины из них диагностируются клинические симптомы СЖ, что связано с низкой пенетрантностью и экспрессивностью варианта.
В крупном российском исследовании показано, что увеличение количества TA-повторов гена ассоциировано с повышением уровня общего и неконьюгированного билирубина, в том числе при носительстве редких аллельных вариантов (TA≤5, TA≥8).
Варианты A(TA)5TAA, A(TA)7TAA и A(TA)8TAA ассоциированы с 130, 65 и 50% активностью фермента УДФ-глюкуронозилтрансферазы 1A1, соответственно, по сравнению с вариантом A(TA)6TAA.
Считается, что снижение экспрессии гена UGT1A1 из-за аномалии в промоторной области является необходимым, но недостаточным изменением для развития клинических проявлений СЖ.
На сегодняшний день СЖ относится к моногенным заболеваниям, развитие которых обусловлено изменениями нуклеотидной последовательности одного гена. При этом наблюдается неполная пенетрантность и вариабельная экспрессивность клинических проявлений варианта rs3064744 гена UGT1A1, что может быть связано с другими молекулярно- генетическими маркерами, которые могут вносить вклад в развитие синдрома.
Таким образом, вероятно, СЖ является не моногенным, а олигогенным синдромом!
2022.A. Ivanova, V. N. Maksimov. Molecular genetic aspects of Gilbert's syndrome, Crigler- Najjar syndromes types I and II // Experimental & clinical gastroenterology, № 204 (8) 2022.
Варианты в гене UGT1A1 связаны с развитием семейной транзиторной гипербилирубинемии новорожденных и метаболизмом ряда лекарственных препаратов.
Так, в случае вариантов UGT1A1*6, rs3064744 гена прием индинавира, иринотекана, атазанавира, сорафениба, тоцилизумаба, белиностата и парацетамола (ацетаминофена) сопряжены с повышенной токсичностью.
rs4148323 (UGT1A1*6)
Вариант rs4148323 (UGT1A1*6) ассоциирован с повышенным риском неонатальной гипербилирубинемии, в гомозиготной форме вариант связан с развитием СЖ.
По данным gnomAD вариант более распространен в Восточной Азии (частота редкого аллеля в азиатской популяции около 0,15, в европейской – около 0,05).
По данным RUSeq частота варианта в российской популяции около 0,014.
Вариант UGT1A1*6 связан со снижением активности фермента УДФ-глюкуронозилтрансферазы 1A1 на 70% по сравнению с диким типом.
В крупном популяционном исследовании в Китае было показано, что с повышенным уровнем общего билирубина ассоциирован промоторный вариант A(TA)7TAA и А аллель варианта UGT1A1*6 гена UGT1A1.
В недавнем исследовании в Китае был проведен молекулярно- генетический анализ промотора и экзонов гена UGT1A1 у 120 лиц с СЖ и 120 здоровых участников группы контроля.
По результатам исследования компаунд-гетерозигот UGT1A1*6 и UGT1A1*28 оказалось больше, чем в исследовании в Индии – 20,83%.
Гомозиготными носителями варианта UGT1A1*28 в группе с СЖ оказались 20%, а варианта UGT1A1*6–15%.
При этом в группе с СЖ уровень общего билирубина был выше у гомозиготных носителей варианта UGT1A1*28, чем у носителей других вариантов нуклеотидной последовательности гена UGT1A1.
2022.A. Ivanova, V. N. Maksimov. Molecular genetic aspects of Gilbert's syndrome, Crigler- Najjar syndromes types I and II // Experimental & clinical gastroenterology, № 204 (8) 2022.

СИМПТОМЫ
- жалобы на тяжесть в правом подреберье
- утомляемость
- ощущение горечи во рту
- тошнота
- головокружение
- желтушные склеры и кожные покровы. (покровы и склеры приобретают желтый цвет при повышении значений билирубина в крови. Данный симптом является наиболее тревожным.)
⠀
АЗИАТСКАЯ МУТАЦИЯ

Азиатская мутация – G71R замена глицина на аргинин (Gly71Arg).
С 2011 года к нам обратились более 4500 человек с подозрением на Синдром Жильбера. Проанализировав статистику и обратную связь от врачей, наша команда начала изучать Азиатскую Мутацию, так как у азиатов мутации обнаруживались в два раза реже, при наличии соответствующей клинической картины.
Валидация метода и тестирование нескольких пациентов с явными симптомами СЖ, у которых не была найдена классическая ТА7-мутация, показало наличие азиатской G71R.
Таким образом, мы добавили азиатскую мутацию наш генетический анализ а Синдром Жильбера.
ПОКАЗАНИЯ
- Подозрение на синдром Жильбера;
- Дифференциальная диагностика синдрома Жильбера, и других заболеваний, проявляющихся гипербилирубинемией;
- Перед началом лечения, с использованием лекарственных препаратов, обладающих гепатотоксическими эффектами;
- Слабовыраженная инфекционная желтуха;
- Хроническая желтушность;
- Увеличение концентрации билирубина при других нормальных биохимических показателях крови;
- Отягощенный семейный анамнез (неинфекционная желтуха, гипербилирубинемия).
⠀
СТАТИСТИКА ЛАБОРАТОРИИ TREEGENE